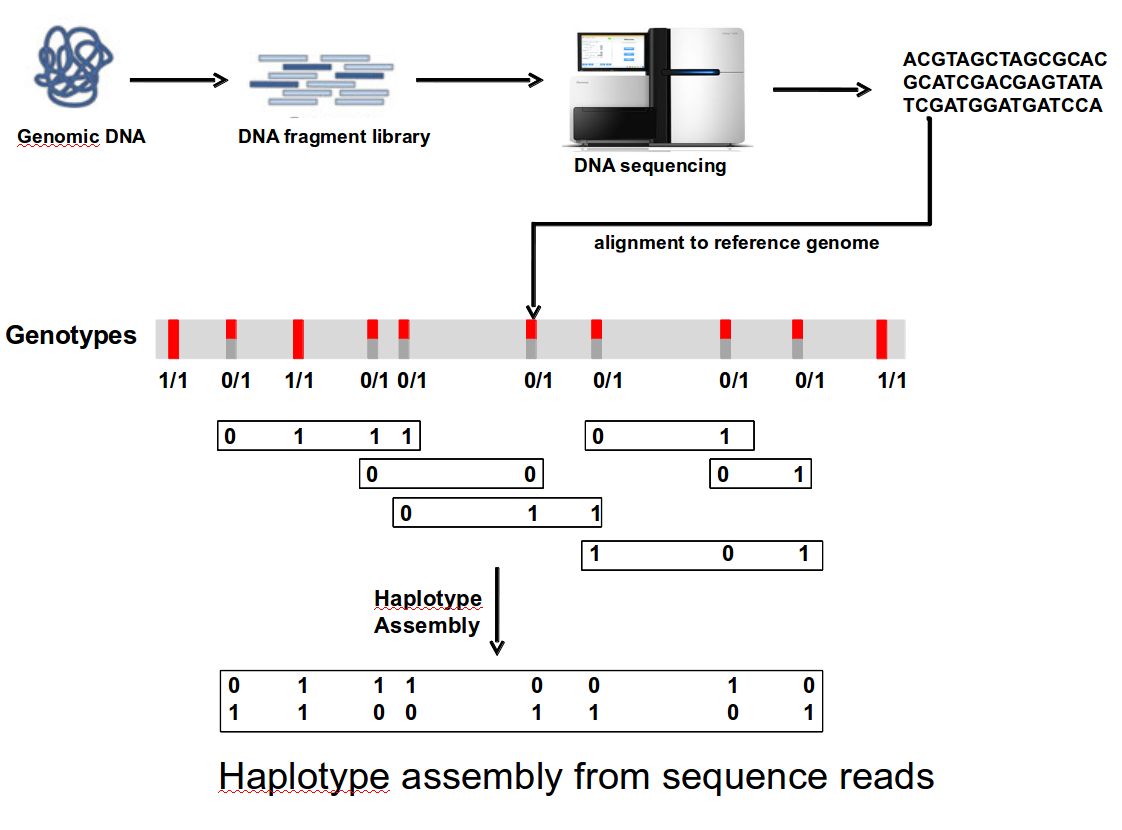
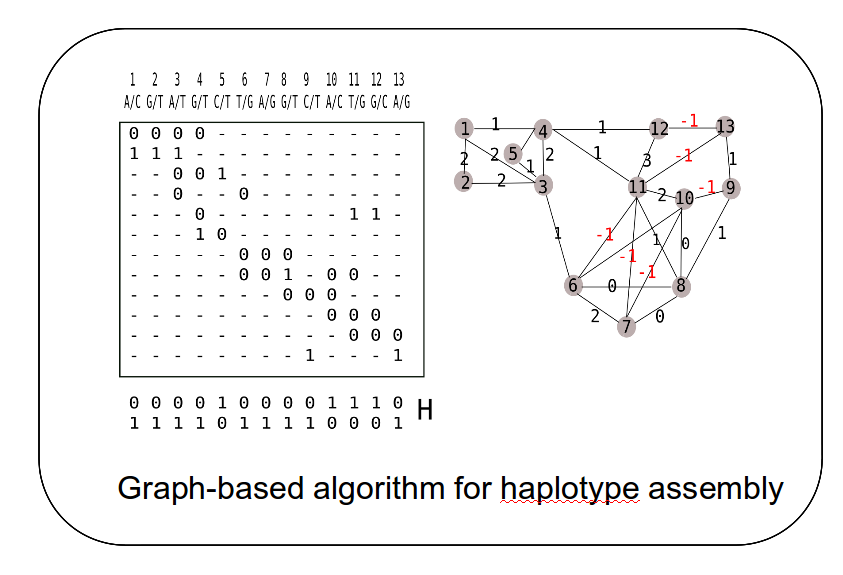
ncing data about human genetic variation is limited in many aspects. In particular, the short read lengths and the short fragment lengths used for genome sequencing limit the recovery of haplotype information. However, many findings indicate that haplotype information (the combination of alleles at variant sites on a single chromosome) is crucial to fully interpret the relationship between human DNA sequence and phenotype including disease. In 2007, we developed the HapCUT algorithm for haplotype assembly from whole-genome sequence data that has been demonstrated to be highly accurate and flexible enough to be applied to a wide variety of data types such as Hi-C data (Selvaraj et al. Nature Biotech 2013). We have published 10 papers on haplotyping in the last 10 years in journals such as Bioinformatics, Genome Research, Nature Biotechnology and Nature Reviews Genetics. Some projects that we are currently interested in: